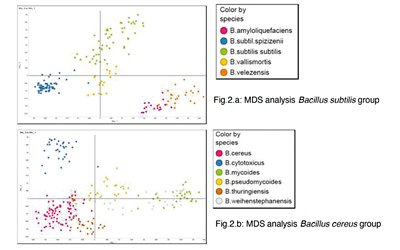
SCIENTIFIC LIBRARY
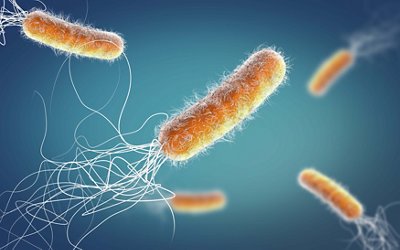
Pseudomonas aeruginosa is a gram negative, rod-shaped bacterium found widely in the environment, such as in soil, water, and plants. They usually do not cause infections in healthy people, but if they do, it is generally mild. It can cause disease in plants and animals, including humans.


?qlt=85&ts=1711540814125&dpr=off)