Overview of sequencing technology

Introduction
Sequencing is not a single technology used for a single purpose. It is a very general term that can be used in many ways. In this article we will discuss the different technologies that all fall under the term sequencing. The aim is toprovide a better understanding of the advantages and disadvantages of these techniques in order to apply them in the correct context. The different technologies can be distinguished by the target of their investigations: either DNA or RNA; either individual strains (bacterial, viral or fungal) or a complete population; either all material present or just specific targeted regions.
All sequencing methods are about determining the order of individual nucleotides A, C, G, T (or U) in a DNA or RNA molecule. RNA sequencing requires some additional preparation steps compared to DNA sequencing. In the final preparation step the RNA is converted to DNA and it is this DNA that is finally sequenced. Therefore, the principle
of RNA sequencing is not different from DNA sequencing and no further distinction between the two will be made in this article.
More and more sequencing innovations have been made in recent years, together with the decreasing cost, this has resulted in a dramatic increase of the applications where sequencing is used. For instance, in public health settings it is now the dominant typing method, and it is even replacing many characterization methods such as serotyping,
virulence profiling and antimicrobial resistance determination.
In recent years, an increasing usage of sequencing technology in microbiology has been observed. The investigation of pathogens is leading the way, but is followed by many other applications, including the research of spoilage organisms. The number of bacterial sequences being deposited in public repositories such as National Center for Biotechnology Information (NCBI), a US public repository of sequences and raw sequencing data is still increasing exponentially.
All sequencing processes following 4 main steps: 1) experimental design, 2) sequencing (wet lab), 3) data management, and 4) data analysis). This article will explore the technologies behind step 2, bacterial sequencing in the lab including some implications for the other steps. A correct experimental design cannot be done without knowledge and understanding of these techniques.

Different Sequencing Technologies
How it works
Bacterial genomes range, on average, from 3 to 5 million base pairs. It would be great to have a technique that could handle this complete DNA molecule, but this is simply not possible yet. So as a first step, the DNA molecule needs to be broken down into smaller pieces to make it more manageable. These smaller pieces, also called reads, will then be sequenced separately, and reassembled using data analysis. Different technologies break the bacterial strain into larger or smaller pieces, but they all follow similar processes.
Traditional sequencing
With the traditional Sanger sequencing, the process required a huge amount of manual work, with each individual fragment needing to be amplified and or purified and placed on the sequencer individually. The method is based on the synthesis of DNA using a small portion of modified nucleotides that contain a colored marker (distinct color per nucleotide) and terminate the synthesis. If this is repeated enough, each position will have one of these modified nucleotides incorporated and you have fragments ending at every position of your sequence colored according to their last nucleotide. If all fragments are now sorted according to size, reading their colors will result in the DNA sequence.
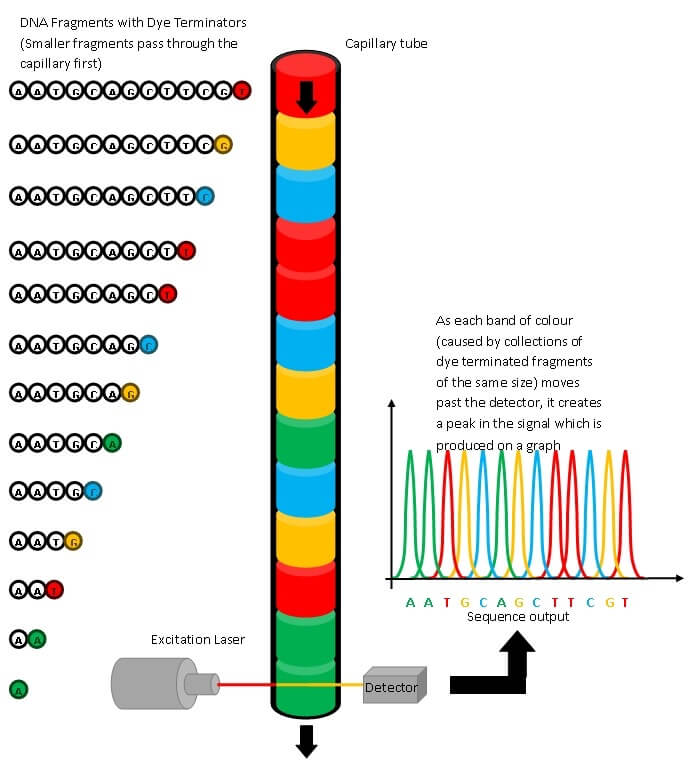 Only fragments of around 500 to 1500 base pairs could be sequenced this way and it also requires a primer sequence that binds to the start of the fragment you are sequencing. For the first genomes, completely sequenced using Sanger, the end of each sequenced fragment was used as the beginning of the next requiring multiple cycles of primer design, amplification, and sequencing. This approach therefore has a high cost and time to result. Next Generation Sequencing (NGS), also called High Throughput Sequencing, has a much higher throughput, and allows for the simultaneous sequencing of many of these smaller pieces. All pieces of a human genome can be sequenced together, for bacterial genomes it is even possible to sequence the pieces of several genomes simultaneously. This dramatically reduces the amount of manual work and therefore the cost.
Only fragments of around 500 to 1500 base pairs could be sequenced this way and it also requires a primer sequence that binds to the start of the fragment you are sequencing. For the first genomes, completely sequenced using Sanger, the end of each sequenced fragment was used as the beginning of the next requiring multiple cycles of primer design, amplification, and sequencing. This approach therefore has a high cost and time to result. Next Generation Sequencing (NGS), also called High Throughput Sequencing, has a much higher throughput, and allows for the simultaneous sequencing of many of these smaller pieces. All pieces of a human genome can be sequenced together, for bacterial genomes it is even possible to sequence the pieces of several genomes simultaneously. This dramatically reduces the amount of manual work and therefore the cost.
Next generation sequencing
In the field of NGS, we make the distinction between two generations of technologies being used today, referred to as the 2nd and 3rd generation (Sanger is considered as the 1st). The two approaches generate data and check for errors in different ways. 3rd generation sequencing is not so much “better” than 2nd generation sequencing as it is different— faster and lower cost — but with more errors. The major difference between the two generations is in the strategies they use to break apart and reassemble the bacterial strain. They both fragment the bacterial genome, make several copies of all fragments, and then sequence everything. The 2nd generation method breaks it into many small segments of 50-400 base pairs. These small pieces are very manageable and can be sequenced with an extremely low error. In 3rd generation, the fragments are much bigger, somewhere between 1500 to 100.000 base pairs, but they are also sequenced with many more errors.
The 2nd generation techniques are still based on DNA synthesis, but instead of a primer binding to the DNA, an adapter is added to the edges of the fragment that binds each fragment in a specific position of a flow cell. All fragments are then amplified at their position, resulting in a cluster of identical fragments. Next, each fragment is copied using marked nucleotides. Images are taken at each incorporation of a nucleotide. The images are of sufficient resolution to detect the color of each individual cluster, allowing to determine the sequence of all fragments bound to the flow cell.
Many different technologies are in use in the 3rd generation, they are based on the detection of a single nucleotide (or the incorporation of a single nucleotide) on a single strand of DNA. The final aim is to be able to obtain the sequence of a complete DNA molecule from just a single organism, but this has not been achieved yet. Some of these methods still use DNA synthesis, but not all. One of the methods without synthesis is based on a protein pore in a membrane that allows the passage of a single strand of a DNA molecule. As the different nucleotides have a distinct size and molecular composition, the configuration of the pore is different for each nucleotide, leading to a distinct disruption of the current through the pore. Measuring these different disruptions allows the determination of which nucleotide is passing through the pore.
When to use which method
Both methods have their advantages and disadvantages for downstream analysis. Puzzling the tens of thousand pieces of a 2nd generation sequencing together is very difficult, especially regions with low complexity (think of the large patch clear blue sky on your 5000-piece puzzle). It is often not possible to make the complete puzzle, you often end up with several large portions of the puzzle that cannot be linked together reliably. These portions are referred to as contigs, the combination of all contigs is called the assembly. The few large fragments you get from a 3rd generation sequencing are much easier to put together to get the complete puzzle, but due to the higher error per base, you will not be able to make out every detail of the puzzle reliably. To sum up, the puzzle will be much more complete with a 3rd generation method, but the details will be easier to distinguish with a 2nd generation method.
Therefore, if details are important, such as determining strain characteristics based on single genes or even single mutations, or for a high-resolution comparison of different strains, a 2nd generation method is most suitable. This is especially the case for a well-known organism where you have good prior knowledge of what the finished puzzle will look like, and which parts are important.  For samples containing multiple organisms, an approach using a 3rd generation method is better suited as the analysis is much more feasible. Keeping with the puzzle metaphor: If you have pieces of ten puzzles mixed together, and all the different images have patches of blue sky, it is nearly impossible to separate them if the puzzles have many small pieces. With a few large pieces, that typically contain some part of the image next to the sky, this is a lot easier.
For samples containing multiple organisms, an approach using a 3rd generation method is better suited as the analysis is much more feasible. Keeping with the puzzle metaphor: If you have pieces of ten puzzles mixed together, and all the different images have patches of blue sky, it is nearly impossible to separate them if the puzzles have many small pieces. With a few large pieces, that typically contain some part of the image next to the sky, this is a lot easier.
For the investigation of populations of microorganisms, also called metagenomics, 3rd generation methods have an advantage.
The detection of viral DNA within a complex sample containing human DNA and bacteria belonging to the human microbiome is also an application for which 3rd generation methods are very popular.
There is one sequencing application that benefits from the joint use of both short-read and long-read methods: the investigation of new species or organisms. If nothing is known about the organism you are sequencing, it helps to have both the overview of the complete puzzle as well as the details of several large patches. The contigs from the 2nd generation methods are placed in their correct position using the information from the 3rd generation method. This is called a ‘hybrid’ assembly.
Different Types of Sequencing
We have now already touched some of the different approaches and applications of sequencing, let us dig a bit deeper. NGS can be used to characterize both an individual isolated strain (referred to as Whole Genome Sequencing here for simplification) as well as a population of multiple organisms (commonly referred to as metagenomics). All DNA in a sample can be sequenced (referred to as shotgun), or only specific DNA sequences can be targeted (referred to as amplicon.) Finally, even though RNA sequencing uses the same techniques as DNA sequencing, it has specific applications that we will briefly discuss.
Single strains versus population
In Whole Genome Sequencing, an attempt is made to characterize all bases of the genome of the investigated strain. This data can be compared to reference databases to learn more about the bacterial strain and make better decisions using this information. Depending on the application, different databases can be used or constructed.
 1. For instance, we can compare the strain to a database with genomes from other strains to see if we have seen the strain before, and where we have seen it before. A food manufacturing site could compare a new Salmonella strain identified to Salmonella strains seen previously at that site to understand if this Salmonella had appeared before, where in the factory, when, and under what conditions. If we have not seen it in the factory, we might be able to compare it to samples from raw materials or lab control strains to identify a potential origin or rule out lab cross-contamination.
1. For instance, we can compare the strain to a database with genomes from other strains to see if we have seen the strain before, and where we have seen it before. A food manufacturing site could compare a new Salmonella strain identified to Salmonella strains seen previously at that site to understand if this Salmonella had appeared before, where in the factory, when, and under what conditions. If we have not seen it in the factory, we might be able to compare it to samples from raw materials or lab control strains to identify a potential origin or rule out lab cross-contamination.
2. We can compare similar strains for degree of difference to understand how closely related they are, and which strain came first. If a factory has seen similar Salmonella strains in the past, we can understand how long the strain has been present based on the degrees of difference (evidence of evolutionary change over time). Strains only mutate if they can grow uninhibited, so looking at the number of mutations in a closely related group of isolates gives us an idea of how well and how long they have been growing in a certain environment. This investigation requires as much information on the strains as possible, when and where they were isolated, how the different isolation locations are related: in the same hygienic zone or not, traffic of vehicles or people between the locations. The insights that can be obtained from such an analysis are very valuable for a root-cause analysis to enable removing the bacteria from its source in the food processing environment and preventing future introduction and spread. As it requires intimate knowledge of the facility and manufacturing process, it can only be done in cooperation with a team from the facility.
 3. We can compare key genetic regions on the strain to databases to identify important characteristics. Target genes can tell us if the strain has already developed the genetic potential for resistance to antibiotics, different industrial cleaning agents or environmental stressors like temperature and moisture. The presence of key genetic structures like plasmids can even tell us how likely a strain is to develop resistance characteristics in the future or to pass it on to other strains.
3. We can compare key genetic regions on the strain to databases to identify important characteristics. Target genes can tell us if the strain has already developed the genetic potential for resistance to antibiotics, different industrial cleaning agents or environmental stressors like temperature and moisture. The presence of key genetic structures like plasmids can even tell us how likely a strain is to develop resistance characteristics in the future or to pass it on to other strains.
In a metagenomics analysis, a snapshot is made of a complete population of microorganisms. Most bacteria do not live as an isolated organism but can form complex populations together with other bacterial species, but also with fungi or viruses. One example is biofilm formation where a more mature biofilm can be formed by several organisms together and some bacteria without the ability to form a biofilm can use a biofilm formed by others as a hide out. It can therefore be particularly useful to investigate the complete population of bacteria and other microorganisms to better understand and anticipate certain risks. In environmental monitoring, it can be used to characterize the populations over time and understand how the composition of this environmental microbiome relates to key product outcomes such as spoilage, taste/texture and potential pathogens. In fermentation it is also very useful as it allows the characterization of the finished product bacterial population in both proportion and quantity to ensure the starter culture is working as expected. During product development of fermented products, it can help identify which mixes drive characteristics like taste, texture and fermentation time. In agriculture and the supply chain, we can monitor both diversity and changes over time to identify important changes that may impact food production.
Shotgun versus amplicon
In shotgun sequencing, all DNA in the sample is fragmented with a random method such as sonication or restriction digestion. All fragments are submitted to sequencing. 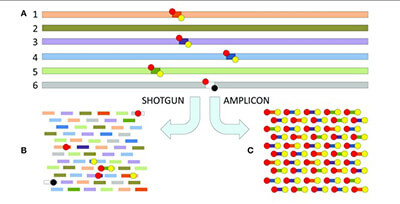 Amplicon sequencing is basically a multiplexed PCR where all PCR products are then sequenced. During analysis the PCR products are separated based on the sequence itself, allowing for a much higher degree of multiplexing than classical multiplex PCR methods that rely on a separation by size and/or colored primer or probe to distinguish between products. With this approach, hundreds to even thousands of targets can be investigated simultaneously.
Amplicon sequencing is basically a multiplexed PCR where all PCR products are then sequenced. During analysis the PCR products are separated based on the sequence itself, allowing for a much higher degree of multiplexing than classical multiplex PCR methods that rely on a separation by size and/or colored primer or probe to distinguish between products. With this approach, hundreds to even thousands of targets can be investigated simultaneously.
In theory, both shotgun and amplicon sequencing can be applied to either DNA from an isolated strain or from a population. In practice, amplicon sequencing is rarely applied to isolated bacterial strains. It is slightly faster than whole genome shotgun sequencing, but this slight improvement on the time to result is rarely worth the effort of selecting the targets and designing the primers. It is easier to simply sequence the complete genome and then extract the targets during analysis. This also has the added flexibility that other targets than initially planned can be extracted. Therefore, in microbiology applications, whole genome shotgun sequencing is simply referred to as whole genome sequencing or WGS. Amplicon sequencing is more common in human genomics, especially cancer genomics where regions important in characterizing the response to chemotherapies are sequenced to decide the appropriate treatment for a patient.
In metagenomics, both shotgun and amplicon sequencing are used. Shotgun sequencing provides a wealth of information, it is a true gold mine of information in which we can keep digging. It is considered as the holy grail of metagenomics. However, it takes a lot of effort to extract any information, the files with the raw data are huge and they require massive computing resources for storage and handling.
Therefore, if the aim of the sequencing can be achieved with amplicon sequencing, this is often preferred. A special application of amplicon sequencing is 16s metagenomics where the targeted sequence is the DNA encoding for the 16s ribosomal unit. This is present in all bacteria and very well preserved within a species and is therefore well suited to identify which bacterial species are in a population. The relative amount of reads from specific 16s sequences is related to the relative abundance of the different species, providing additional information on the population structure. For fungal organisms, a region called ITS which is in between the DNA encoding for the different ribosomal subunits is a more suitable target.
RNA sequencing
As RNA is converted to DNA before sequencing, it can be sequenced using the same technologies. RNA sequencing is mainly applied to the investigation of RNA viruses and transcriptomics.
RNA viruses are typically quite small, in the order of a few thousand base pairs. This is within the range of fragments of third generation sequencing methods. As a result, many viral genomes can be reconstructed on a single read. Another advantage of certain 3rd generation methods is they do not require the isolation of the virus or large concentrations of viral RNA, which is both very difficult to achieve as virus replication requires a host cell. For detection of the absence or presence of a virus in a sample, 3rd generation methods are very powerful. Once the genome of one strain has been well described, no isolation from the host cells is needed for variant detection, the reads are simply compared to the known genome. Both 2nd and 3rd generation methods are suitable for this.
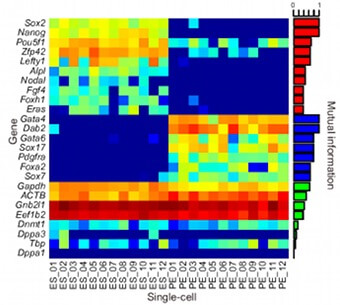 Transcriptomics is the investigation of the RNA transcripts present in an organism at a specific moment. When performed with a high throughput sequencing method, it is also called RNA-seq. Where the presence of certain genes in the genome describes the potential of a characteristic, the presence of a transcript confirms this potential. A transcript of a gene is only made when the corresponding protein is under production, the more transcripts of a gene are present, the more protein is being made. So not just the presence of certain transcripts, but also their copy number provides information on the complete transcriptome of an organism under certain conditions. Most researchers prefer 2nd generation methods, as they typically sequence more copies of the original RNA, resulting in better confidence in the relative concentrations and thus expression levels of the genes. However, the ability to sequence the transcripts end-to-end with 3rd generation methods has lately been convincing many researchers to convert to these technologies.
Transcriptomics is the investigation of the RNA transcripts present in an organism at a specific moment. When performed with a high throughput sequencing method, it is also called RNA-seq. Where the presence of certain genes in the genome describes the potential of a characteristic, the presence of a transcript confirms this potential. A transcript of a gene is only made when the corresponding protein is under production, the more transcripts of a gene are present, the more protein is being made. So not just the presence of certain transcripts, but also their copy number provides information on the complete transcriptome of an organism under certain conditions. Most researchers prefer 2nd generation methods, as they typically sequence more copies of the original RNA, resulting in better confidence in the relative concentrations and thus expression levels of the genes. However, the ability to sequence the transcripts end-to-end with 3rd generation methods has lately been convincing many researchers to convert to these technologies.
Different Sequencing Technologies
To make a good decision on the most suitable method, the aim of your experiment needs to be defined very well. Once this is done, the decision is likely very clear. One final major point of consideration to make when deciding on the sequencing technology is cost. The pricing of different methodologies is constantly changing. For most applications, 2nd generation sequencing methods are still cheaper, but the difference is growing smaller. For all techniques, a major factor in the price is throughput. A machine with a higher capacity has a lower cost per sequenced Gbp, but only if the capacity is used to its max. If the machine is run at lower capacity, the price gets increasingly higher. If the time to result is not important, you can simply wait until you have enough samples to make a complete run. For some applications however, you want the results as soon as possible. For these applications, it is more cost effective to use a lower capacity machine at full capacity rather than constantly running a high capacity machine at a fraction of its maximum. Therefore, it is very important to consider the amount of samples you need to sequence and the time to result that is required during experimental design. If you ever wondered why sequencing just a few samples with a short turnaround time in a commercial sequencing lab is so much more expensive than a large batch, this also answers that question.